
En sólo dos años el Fisco ha autorizado el pago $7.500 millones por trato directo a Medtronic, el único proveedor de bombas de insulina bajo la Ley Ricarte Soto. Aunque la bomba mejora la calidad de vida de los pacientes, es el dispositivo médico que registra más reportes de fallas en Estados Unidos en la última década. Medtronic es la empresa de dispositivos médicos más grande del mundo y también la más cuestionada en la investigación internacional The Implant Files. En Chile, le ha vendido $133 mil millones al Estado desde 2015.
Que el futbolista Arturo Vidal se haga otro tatuaje está lejos de ser noticia, pero el que mostró a mediados de 2016 lo fue. Era distinto a las decenas de ilustraciones que cubren todo su cuerpo: en el costado derecho de su abdomen, sobre la ingle, el “rey” se tatuó una bomba de insulina. Se trata del dispositivo médico que usa su hijo Alonso para mantener a raya la diabetes tipo I. Con su tatuaje, Vidal buscaba convencer al gobierno chileno de que comprara estos carísimos dispositivos médicos para todos los niños que padecen esa enfermedad.
Vidal fue parte de una exitosa campaña que consiguió que en 2017 la bomba de insulina se incluyera dentro de la llamada Ley Ricarte Soto, que dispone el financiamiento estatal para el tratamiento de enfermedades de alto costo.

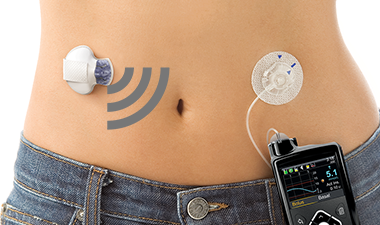
La bomba de insulina genera una mejora importante en la calidad de vida de las personas con diabetes tipo 1. Les evita pincharse constantemente para chequear su nivel de glucosa, pues lo mide de manera continua. Tampoco es necesario usar inyecciones para aplicarse insulina, ya que la suministra directamente. El dispositivo −bien usado y si funciona sin fallas− debería prevenir episodios glicémicos graves.
Por eso el Estado decidió financiarlo para niños y adultos. En mayo de 2017 autorizó la compra de 846 bombas de insulina y sus respectivos kits de insumos. Aunque según Medtronic sólo se han entregado 650 aparatos, en mayo de 2018 una nueva resolución autorizó la compra de otros 846 dispositivos. En total, se ha aprobado el pago de $7.500 millones (US$11 millones) en bombas de insulina Minimed 640G, más sus insumos.
Las compras se han hecho de manera directa, sin licitación, bajo el argumento de que Medtronic es el único proveedor de bombas de insulina de este tipo. Los expertos consultados confirman que si bien hay bombas de otras marcas, éstas no incluyen el monitoreo continuo de glucosa ni la suspensión automática de insulina. El producto de Medtronic es el más completo.
El precio de mercado de una Minimed en Chile supera los $4,6 millones de pesos, a lo que hay que sumar la compra permanente de insumos cuyo costo mensual es de más de $350 mil. Eso ponía al dispositivo fuera del alcance de la gran mayoría de los enfermos. El contrato con la Cenabast, sin embargo, establece que cada bomba se vendió a $1,9 millones (menos de la mitad que en el mercado), claro que la empresa se aseguró 1.700 pacientes cautivos a quienes el Estado financiará mensualmente sus insumos (vea la resolución que autoriza la compra directa a Medtronic y la que renueva el contrato).
El rol de Vidal fue sólo una pieza de la fuerte campaña para que el Ministerio de Salud incorporara la bomba de insulina entre las prestaciones gratuitas. En 2015, las organizaciones de pacientes reunieron 42 mil firmas de apoyo. Entre quienes lideraron la campaña estuvieron el ex tenista Hermes Gamonal y Marcelo González, autor del sitio midiabetes.cl y a quien Medtronic eligió para testear su nueva bomba en Chile. Ambos se reunieron en octubre del mismo año con el director del ISP, Alex Figueroa, y en mayo de 2016 con la entonces directora de Fonasa, Jeanette Vega. En paralelo, la empresa realizó lobby directo ante las autoridades del Minsal.
Tanto Medtronic como su producto estrella Minimed ocupan un lugar protagónico en la investigación “The Implant Files”, liderada por el Consorcio Internacional de Periodistas de Investigación (ICIJ por su sigla en inglés), en la que participaron 252 periodistas de 36 países, y de la que LaBot es parte.
La investigación de la Dutch Public Broadcasting reveló cómo una malla de mandarinas pudo llegar al mercado como implante vaginal, reflejando la falta de regulación sobre dispositivos médicos.
La multinacional basada en Minnesota, pero con domicilio en Dublín, es la mayor fabricante de dispositivos médicos del mundo y también una de las más cuestionadas, pues encarna gran parte de los vicios de una industria que tiene el potencial de mejorar la vida de las personas. Pagos indebidos a médicos, lobby sin límite ante autoridades y avisos de fallos fuera de tiempo que ponen en riesgo la vida de los pacientes, son algunos de los cuestionamientos detectados por ICIJ (ver reportaje en español). En Chile, según un análisis de las órdenes de compra de la empresa y sus filiales, sólo en los últimos tres años el Estado le ha comprado $133 mil millones (casi US$200 millones) a Medtronic en distintos tipos de dispositivos médicos y servicios clínicos..
Aunque la tecnología de sus bombas de insulina ha significado un avance para el tratamiento de la diabetes, al mismo tiempo carga con un complejo historial. En un país como Chile, donde este tipo de dispositivos no cuenta con regulación, la millonaria inversión en un producto como Minimed debería al menos encender una luz de preocupación.
LOS CUESTIONAMIENTOS A MINIMED
Maude es el nombre la base de datos de la Food & Drug Administration (FDA, regulador estadounidense) que registra los reportes de empresas, médicos y pacientes sobre problemas en los dispositivos médicos. Entre los cientos de miles de avisos que Maude recibe cada año, los dispositivos que más se repiten en la última década (2008-2017) son las bombas de insulina.
El análisis que hizo ICIJ junto a la agencia Associated Press de esos datos arroja que en ese periodo las bombas de insulina son los dispositivos con más reportes de eventos adversos. Las de Medtronic, en específico, están potencialmente asociadas a 150 mil lesiones de pacientes. En el mismo periodo, hubo 20 retiros de mercado (o recallsen inglés) de las bombas de insulina de Medtronic y se interpusieron cerca de 100 acciones judiciales por su mal funcionamiento.

En su respuesta a ICIJ, Medtronic relativizó esos números, afirmando que las comunicaciones recibidas por la FDA pueden estar basadas en información incompleta o engañosa. Las bombas de insulina, argumentó la empresa, han ayudado a cientos de miles de diabéticos, y el retiro o recall de un dispositivo no significa que sea peligroso.
Más allá de las interpretaciones, es un hecho que las bombas de insulina de Medtronic arrastran una historia de fallos. Uno de los más graves ocurrió en julio de 2013, cuando la empresa notificó a la FDA de problemas con el equipo de infusión de su bomba de insulina Minimed Paradigm y envió un recall de los infusores del equipo, advirtiendo que podrían suministrar cantidades equivocadas de insulina. El episodio fue tan riesgoso para la salud de las personas que la FDA lo calificó como Clase I, es decir, de aquellos que pueden provocar grave daño o incluso causar la muerte de un paciente.
Poco después, en septiembre del mismo año, la FDA realizó una visita inspectiva a los laboratorios de Medtronic como parte del proceso de aprobación de un nuevo modelo de bombas de insulina. El resultado fue una dura carta de advertencia a la empresa, por no cumplir con los estándares de buenas prácticas exigidos por el órgano regulador.
Según ICIJ, la FDA amonestó a Medtronic por no investigar o responder a tiempo a las reclamaciones de los consumidores o incluso a las muertes de pacientes. El ente regulador incluso alertó que en un caso el aviso tardío sobre el posible mal funcionamiento de una bomba podría haber provocado una sobredosis de insulina, lo que a un paciente le originó un coma diabético. Además, la FDA hacía ver que las bombas de Medtronic no cumplían las regulaciones de calidad en aspectos como fugas de insulina o fallos del software.
La base de datos de la FDA arroja que en siete de los retiros de bombas de insulina de Medtronic figuran dispositivos que también fueron distribuidos en Chile. Sin embargo, no hay ningún registro público en el país que indique que los pacientes fueron alertados de los problemas que presentaban los dispositivos.
Algunas alertas son simplemente “mundiales”, como sucedió en septiembre del año pasado, cuando Medtronic hizo un amplio llamado para que sus clientes revisaran los sets de infusión de sus bombas de insulina –el dispositivo que suministra el fármaco al organismo– estaban entre los lotes afectados:
“Medtronic ha sido alertada de reportes recientes de potenciales excesos de la entrega de insulina poco después del recambio de set de infusión. El exceso de insulina puede causar hipoglicemia y en casos extremos, la muerte. Medtronic ha recibido reportes de hipoglicemia que han requerido atención médica y que están potencialmente relacionados con este problema”.
Ese retiro voluntario ocurrió cuatro meses después de que se firmara la resolución autorizando la compra de bombas de insulina por parte de la Cenabast.
Si en Estados Unidos la fiscalización que hace la FDA de los dispositivos médicos es seriamente cuestionada, en Chile simplemente no existe y mucho menos hay un registro público con las alertas de fallos. Sin embargo, la Cenabast, que compra y distribuye las bombas de insulina entre los beneficiarios de la Ley Ricarte Soto, actualiza periódicamente una planilla con los reclamos de los pacientes que utilizan sus insumos. Minimed es por lejos el dispositivo que más reclamos tiene (50 de los 115 recibidos entre enero y septiembre). Ahí se mezclan problemas que son responsabilidad de los usuarios, como pérdida y rotura de equipos, con fallas de calidad que obligan a cambiar el producto.
Aunque los ejecutivos de Medtronic en Chile no accedieron a dar una entrevista, la gerenta de comunicaciones, Sandra Ocampo, respondió por escrito que “las bombas de insulina son dispositivos importantes y que cambian la vida de muchos pacientes con diabetes” y que de las 650 bombas entregadas a la Cenabast, 57 han sido reportadas (8,7%). Sin embargo, agregó que sólo el 3% del total (equivale a 19,5 dispositivos) corresponde a algún tipo de falla. Todas fueron reemplazadas en 24 horas, aseguró.
Las fallas individuales se suman a las alertas internacionales. La última tuvo lugar el 10 de octubre pasado, cuandoMedtronic notificó que había recibido reportes de fallas en las bombas de insulina Minimed 640 G –las mismas que Cenabast distribuye gratuitamente a los enfermos en Chile–, respecto a la alarma que avisa si los niveles de glucosa están demasiado altos o demasiado bajos. En las páginas web locales de la empresa se explicaba cómo chequear si el aparato tenía problemas y, en caso de que los tuviera, advertía que debía ser reemplazado para evitar el riesgo de hipo o hiperglicemia.
Pese a la alerta, Medtronic recalcó que “los eventos adversos se informan para todos los dispositivos y productos médicos, y no indican en sí mismos que existe un problema de seguridad. Como ha dicho la FDA: el número de informes no se puede interpretar o utilizar de forma aislada para llegar a conclusiones sobre la existencia, gravedad o frecuencia de los problemas asociados con los dispositivos«.
Según Medtronic, en Chile los pacientes fueron alertados del problema por email o por teléfono, y a través de Cenabast, que es el protocolo acordado con el Ministerio de Salud. No fue posible confirmar los detalles con la Cenabast, pues sus directivos, al igual que los de Medtronic, se negaron a dar una entrevista para este reportaje.

Misty Holliman vive en Texas. No fue informada de los riesgos de Essure. (Foto: Can Turkyilmaz / ICIJ).
En mayo de 2018, a Misty Holliman, de 26 años, una madre de cuatro hijos que vive en las afueras de Irving, Texas, se le implantó Essure. Ella es una de más de 200 pacientes que usaron dispositivos médicos y que informaron a ICIJ o a uno de sus colaboradores que no estaban informadas de los riesgos para su salud antes de que les colocaran el dispositivo.
Poco tiempo después, en julio, Bayer anunció que dejaría de vender ese dispositivo en Estados Unidos a finales de año. En sus comentarios a ICIJ, Bayer dijo que su decisión de retirar los dispositivos a nivel mundial está motivada por razones comerciales más que por preocupaciones por la seguridad del producto, y citó una disminución general en el uso de anticonceptivos permanentes, así como “publicidad inexacta y engañosa sobre el dispositivo”. Bayer también señaló que las mujeres que han demandado a la compañía han generado muchos de los informes de incidentes adversos presentados a la FDA.
Holliman ahora sufre de dolor pélvico severo. Es posible que necesite una histerectomía completa y no puede pagar el procedimiento. “No puedo ver lo que pasa dentro de mi cuerpo”, lamentó, “y no puedo sacarlo”.
UNA REGULACIÓN TARDÍA
Los modernos requisitos de pruebas para nuevos medicamentos se establecieron a raíz de un escándalo médico que sacudió al mundo. A fines de la década de 1950 y principios de la de 1960, la talidomida, un medicamento administrado a las mujeres para tratar las náuseas matutinas, hizo que decenas de miles de niños nacieran con extremidades malformadas y una amplia variedad de otros defectos congénitos. Hasta 40% de los bebés con exposición significativa a ese medicamento murieron en la infancia y muchas mujeres experimentaron mortinatos y abortos espontáneos.
En respuesta, surgió una avalancha de nuevas regulaciones farmacéuticas. A pesar de los enormes costos asociados, se exigió a los fabricantes de medicamentos que presentaran pruebas clínicas que demostraran que sus productos eran seguros y eficaces antes de que pudieran salir al mercado.
La regulación de la industria de los dispositivos fue dejada como un tema de última hora y evadió la supervisión total hasta 1976 en Estados Unidos y 1990 en Europa. La industria argumentó desde el principio que sus dispositivos deberían ser tratados de manera diferente a los medicamentos.
El criterio aceptado internacionalmente para aprobar casi todos los nuevos medicamentos es que al menos un ensayo controlado aleatorio, en pruebas realizadas con humanos, debe demostrar seguridad y eficacia. En cambio, el estándar para la aprobación de nuevos dispositivos es más bajo en todo el mundo.
En EE.UU. los fabricantes de medicamentos deben exhibir “evidencia sustancial” de la seguridad y efectividad de un nuevo producto y, por lo general, requieren tres ensayos. Para los dispositivos, el punto de referencia es la “seguridad razonable”, que generalmente significa un único estudio y ningún ensayo controlado aleatorio, en el que los grupos de pacientes reciben diferentes tratamientos y se comparan los resultados.
Pero incluso este estándar no siempre se pone en práctica. Menos de 5% de los dispositivos revisados por la FDA se someten a la llamada “aprobación previa” a la comercialización. Los reguladores permiten cambios importantes —a veces fatales— en los dispositivos, realizados por las vías destinadas a las actualizaciones incrementales.
La mayoría de los dispositivos se aprueban si son “sustancialmente equivalentes” a los dispositivos que ya están en el mercado o a una versión anterior del mismo producto.
En algunos casos, después de una cadena de aprobaciones basadas en equivalencias, un nuevo dispositivo apenas si se parece a la versión original. La investigación publicada por la revista médica BMJ, socia de ICIJ en este proyecto, rastreó el árbol genealógico de 61 productos de malla quirúrgica y al final llegó hasta dos dispositivos aprobados en 1985 y 1996. Ninguno había completado los ensayos clínicos al momento de ser aprobados.
Los pacientes que toman medicamentos de mala calidad pueden deshacerse de ellos sólo con tirar el frasco de pastillas a la basura, señaló Adriane Fugh-Berman, profesora de la Universidad de Georgetown que estudia las prácticas de marketing de la atención médica. Las personas con un implante innecesario o que funciona mal pueden terminar llevándolo dentro de su cuerpo por el resto de sus vidas. “Puedes quedar lisiado para siempre”, dijo.
FALLA DEL DESFIBRILADOR: “GOLPEADA POR UN RAYO”
Medtronic obtuvo en 2004 la aprobación de una versión actualizada de un cable utilizado para conectar su desfibrilador implantable al corazón. Llamado Sprint Fidelis, el cable era mucho más delgado que las versiones anteriores, una innovación que se consideró una ventaja porque los alambres finos son más dúctiles y más fáciles de doblar.
Durante los tres años siguientes, el Sprint Fidelis se implantó en 268 mil pacientes en el mundo.
En enero de 2007, Sherry Robinson, de 32 años de edad, se preparaba para acostarse en su casa de Sechelt, una comunidad costera en las afueras de Vancouver, Canadá, cuando una impactante sacudida en el pecho la empujó hacia adelante. “Vi una luz blanca a través de mis ojos. Dolió como el demonio. Pensé: ‘Me ha caído un rayo’”.
El dispositivo que lleva Robinson está diseñado para que su corazón recupere el ritmo, pero las pruebas de hospital mostraron que estaba fallando. Antes de ser desactivado, un cable Sprint Fidelis defectuoso electrocutó 18 veces a Robinson.
“Casi nadie puede tolerar descargas eléctricas múltiples”, comentó un cardiólogo citado en un informe del Servicio de Investigación del Congreso publicado casi una década después. “Después de una segunda o tercera descarga, la ansiedad causada por la posibilidad de más descargas se convierte rápidamente en terror”.
Los médicos retiraron ese mismo mes el dispositivo defectuoso que llevaba Robinson —otra cirugía—, pero luego simplemente colocaron otro Sprint Fidelis.
En julio de 2007, un cardiólogo de Minnesota publicó un estudio que mostraba que el Sprint Fidelis fracasó a una tasa más alta de la esperada y que estaba dando descargas eléctricas a los pacientes o fallando.
(Foto: Robina Weermeijer on Unsplash)
Citando la muerte de cinco pacientes que pudieron estar relacionadas con un cable fracturado de Sprint Fidelis, la compañía retiró el dispositivo y la sacó del mercado.
Dos años después, Medtronic reconoció que el mal funcionamiento de Sprint Fidelis puede haber causado 13 muertes, aunque no quedó claro si éstas se sumaban a las cinco mencionadas por la compañía cuando anunció el retiro del dispositivo.
Su rendimiento se deterioró con el tiempo. Un estudio realizado en 2015 con cerca de mil pacientes en Francia encontró que, de hecho, más de uno de cada cinco cables se fracturaron después de cinco años. Los pacientes más jóvenes y activos eran especialmente vulnerables.
Un análisis de ICIJ de los informes de incidentes adversos de la FDA muestra que en la década pasada los incidentes adversos vincularon a varios modelos de Sprint Fidelis con más de 8 mil lesiones y 2 mil muertes.
Aunque la FDA consideraba al Sprint Fidelis como un dispositivo de alto riesgo, éste no fue sometido a pruebas con pacientes: la agencia lo aprobó a través de un “suplemento” a una versión que había sido autorizada más de una década antes.
Medtronic no respondió a una pregunta específica sobre Sprint Fidelis, pero dijo que no lanza un dispositivo o terapia al mercado “a menos que y hasta que hayamos confirmado que el producto es seguro y eficaz en el tratamiento de la condición médica en cuestión”. La compañía también comentó que continúa monitoreando la seguridad y el rendimiento de sus dispositivos una vez que se encuentran en el mercado.
APROBACIONES EXPEDITAS
Las nuevas versiones de dispositivos de alto riesgo también han llegado al mercado a través de una vía expedita de equivalencia sustancial llamada 510(k). En 2009, un reporte de la Oficina de Responsabilidad Gubernamental (Government Accountability Office) de EE.UU. criticó a la FDA por continuar aprobando dispositivos de alto riesgo por medio del programa 510(k) a pesar de que el Congreso hacía décadas que había ordenado no hacerlo.
Dos años después, el Instituto de Medicina, hoy una unidad de las academias nacionales de Ciencias, Ingeniería y Medicina, instó a la FDA a eliminar el programa 510(k) por completo. La FDA rechazó la recomendación por considerarla inviable.
Los dispositivos de alto riesgo siguen saliendo al mercado a través de la laguna 510(k) —al menos 42 de ellos desde 2011—, según encontró ICIJ. Seis de ellos, incluidos varios desfibriladores y sus monitores, posteriormente fueron objeto de 14 retiros del mercado.
Janice Hogan, abogada de la industria de dispositivos entrenada en ingeniería biomecánica, advierte que aunque algunas solicitudes presentadas al programa 510(k) son relativamente sencillas, muchas incluyen pruebas extensas en humanos y miles de páginas de documentación. “La FDA tiene una discrecionalidad sustancial sobre los datos que se requieren”.
En las respuestas escritas a las preguntas planteadas por el ICIJ, la FDA dijo que no aprobó ningún dispositivo de alto riesgo a través de 510(k) en 2017.
“Durante los últimos años, la FDA ha hecho un esfuerzo concertado para asegurar que se exija el nivel apropiado de pruebas”, dijo la agencia. En algunos casos, la FDA puede exigir “pruebas exhaustivas” para los dispositivos sujetos al programa 510(k), lo que requiere más evidencia, señaló la agencia en respuestas escritas a las preguntas de ICIJ.
Las pruebas clínicas y los ensayos con pacientes no son apropiados ni necesarios para la mayoría de los dispositivos, según la FDA. Eliminar las aprobaciones expeditas “no necesariamente proporcionaría mejores salvaguardas para los pacientes, sino que provocaría costos y retrasos innecesarios, a la vez que desviaría los recursos del personal de la FDA del estudio y la evaluación de dispositivos de mayor riesgo y novedosos”, señaló la organización.
Trunzo, portavoz de AdvaMed, cuestionó la noción de que se necesitan ensayos clínicos sólidos para la aprobación de dispositivos, y argumentó que otras formas de pruebas no clínicas pueden producir conclusiones más precisas.
LA VENTAJA EUROPEA
El sistema de aprobación de implantes de la Unión Europea es aún más favorable para las empresas. De hecho, es un negocio.
Los fabricantes de dispositivos pagan a empresas privadas, conocidas como organismos notificados, para que certifiquen que los dispositivos de riesgo alto y medio cumplen las normas de seguridad europeas.
Los grandes actores, entre ellos el Grupo BSI en el Reino Unido y las empresas alemanas TUV Rheinland y TUV Sud, marcan los dispositivos médicos con el mismo sello de CE que aparece en muchos productos de consumo en Europa, como tostadoras, fuegos artificiales, juguetes para niños, etc., y certifica que cumplen con los “requisitos esenciales” de seguridad y protección del ambiente.
Entre las ventajas para la industria, la mayoría de los organismos notificados están exentos de cumplir las leyes que requieren que las agencias gubernamentales hagan públicos los registros de las aprobaciones de dispositivos. En el caso de los implantes, esto es especialmente preocupante.
Según un correo electrónico de marzo de 2016 entre los principales funcionarios de la salud de Alemania y Dinamarca, los reguladores de la UE no cuentan con datos clínicos de 90% de los dispositivos de mayor riesgo porque fueron evaluados como suficientemente similares a los productos existentes.
Los defensores de los pacientes han luchado durante mucho tiempo para eliminar el sistema de organismos notificados, llamándolo opaco, profundamente conflictivo y propenso a permitir que aparezcan dispositivos desastrosos en el mercado. Aun así, la etiqueta CE es aceptada en todo el mundo, lo que convierte a Europa en un importante punto de entrada para la industria de dispositivos. En Arabia Saudita, India, Filipinas, Singapur y gran parte de América Latina, los dispositivos son bienvenidos o sometidos a menos escrutinio si ya han sido certificados en Europa.
MENOS BARRERAS, MÁS MERCADO
Los fabricantes de dispositivos compiten para llevar nuevos dispositivos al mercado e introducir modelos con nuevas características para mejorar la calidad e impulsar las ventas. Los analistas de Wall Street siguen de cerca el tiempo que toma superar los obstáculos regulatorios. Los expertos estiman que el ciclo de vida estándar de un dispositivo antes de ser reemplazado por el siguiente modelo es ahora de sólo 18 a 24 meses.
Y a medida que la industria crece, ocurre lo mismo con la complejidad de sus dispositivos.
Boston Scientific Corp., Medtronic, Abbott Laboratories y otros fabricantes de dispositivos venden “estimuladores del nervio vago”, implantes que envían impulsos eléctricos al cerebro, cuello o abdomen para combatir dolencias que van desde dolor de espalda hasta hipo crónico y depresión. Las válvulas cardiacas plegables, fabricadas por Edwards Lifesciences y otras compañías, pueden ser colocadas dentro del corazón a través de una pequeña incisión usando catéteres orientables, que se abren por completo al llegar a su destino, como un barco en una botella. Medtronic fabrica una “bomba para el dolor” implantable que envía microdosis de fármacos analgésicos a la columna vertebral y genera informes de rendimiento que los pacientes pueden leer en un tablet. Estos dispositivos proporcionan beneficios, pero también conllevan riesgos.
A veces los cálculos de riesgo-recompensa pueden ser exquisitamente minuciosos. Por ejemplo, la válvula cardiaca plegable, llamada reemplazo de válvula aórtica transcatéter (TAVR) en Estados Unidos, es conveniente para los ancianos o enfermos porque elimina la necesidad de realizar una traumática cirugía a corazón abierto. Pero nadie sabe cuánto tiempo duran, por lo que tiene menos sentido cuanto más joven y saludable sea el paciente. Pero, ¿qué tan joven y saludable?
Ser la primera en comercializar un producto pionero puede asegurarle un éxito enorme a una empresa. En 2014, Dan Starks, entonces director ejecutivo de St. Jude Medical Inc., apareció en Mad Money, un programa de televisión de la CNBC. Sacó de su bolsillo un implante más pequeño que una pila AAA y lo sostuvo en alto mientras la cámara se acercaba a él.
Se trataba del Nanostim, el primer marcapasos inalámbrico, sin los delgados cables que conectaban el dispositivo al corazón, que durante mucho tiempo habían causado problemas a los fabricantes. “Esto va a revolucionar el campo”, dijo Starks.
El diseño sin cables de Nanostim fue un importante atractivo de venta. Las fracturas u otros problemas de funcionamiento de los cables, como el Sprint Fidelis, habían afectado a los dispositivos cardiacos anteriores.
“Puedo decirles que se realizó el primer implante con esta tecnología en Gran Bretaña justo en esta última semana, y el tiempo del procedimiento de implante en manos de ese médico en particular fue de ocho minutos”, alardeó Stark en Mad Money.
(Foto: Piron Guillaume on Unsplash)
La paciente era Maureen McCleave, una abuela de 77 años, oriunda de Londres. “Me siento como si fuera una mujer nueva”, comentó al periódico Daily Mail en una de las varias entrevistas concertadas por St. Jude poco después de salir del hospital. “Si me hubieran puesto un marcapasos tradicional, probablemente seguiría en el hospital y no me sentiría tan bien como ahora”.
Tres meses después de la operación, St. Jude reveló las primeras preocupaciones sobre el Nanostim. Los médicos habían descubierto que seis de cada 147 pacientes que participaron en un ensayo europeo habían sufrido una perforación del músculo cardiaco. Dos habían muerto. Más tarde, la batería del Nanostim falló en varios otros pacientes, haciendo inútiles sus marcapasos.
Los problemas con la batería se hicieron tan frecuentes que St. Jude pidió en 2016 a los médicos que utilizaban el Nanostim que hicieran una “pausa” y dejaran de implantar estos dispositivos. Esa pausa ha durado desde entonces.
A finales de 2016, la abuela londinense comenzó a tener palpitaciones cardiacas y a sentirse cansada. “Sabía que algo andaba mal en alguna parte”, confesó McCleave a los socios de ICIJ.
Una enfermera le explicó que su marcapasos se había detenido. McCleave, que entonces tenía 80 años, necesitaba urgentemente otra cirugía. Aunque se había asegurado que la batería del Nanostim duraría hasta 19 años (seis años más que un marcapasos estándar), el de McCleave había fallado en tres.
Los marcapasos tradicionales son relativamente fáciles de reemplazar una vez que la batería se agota. Se colocan justo bajo la piel, debajo de la clavícula, y envían impulsos eléctricos al corazón a través de cables que pueden permanecer en su lugar si requieren ser reemplazados. Extraer un Nanostim del interior del corazón es mucho más complejo.
La segunda operación de McCleave fue difícil y complicada. Los cirujanos implantaron otro marcapasos y dejaron el Nanostim fallido en su lugar, al juzgar que era demasiado peligroso retirarlo. “Fue muy difícil porque sangré mucho”, recordó McCleave.
El implante de corazón futurista de St. Jude había sido certificado y su seguridad había sido avalada con un mínimo de pruebas. La Unión Europea exige que incluso los medicamentos más comunes se sometan a al menos un ensayo clínico en cientos o miles de pacientes para determinar su seguridad y eficacia. El Nanostim había sido probado en 33 humanos y durante un tiempo relativamente corto, de 90 días. Los únicos otros sujetos de prueba fueron 30 ovejas.
McCleave dijo que nadie de St. Jude, ahora parte de Laboratorios Abbott, le explicó por qué su Nanostim había fallado. “Me sentí como un pedazo de basura que había sido arrojado a un lado”, recordó McCleave.
EL CABILDEO CAMBIA EL PANORAMA EN EE.UU.
Los reguladores de EE.UU. han criticado sin rodeos la regulación de los dispositivos médicos en el resto del mundo en general, y en Europa en particular.
“No usamos a nuestra gente como conejillos de indias en Estados Unidos”, sostuvo el jefe de dispositivos de la FDA, el médico Jeffrey Shuren, en una llamada con reporteros en 2011 durante un debate en el Congreso sobre la conveniencia de adoptar más normas de estilo europeo en Estados Unidos. El comentario provocó un alboroto diplomático.
La FDA emitió en 2012 un informe que llamaba por su nombre a los “dispositivos inseguros e ineficaces” aprobados con unas pocas pruebas en la Unión Europea. La lista incluye endoprótesis utilizadas para reparar aneurismas o abombamientos en las paredes aórticas. La FDA encontró que muchos presentaban “riesgos graves para los pacientes, incluyendo coágulos de sangre, fallas del injerto y ruptura de aneurismas”.
Al año siguiente, la FDA emitió un mensaje diferente: Estados Unidos se esforzará por ser “el primero en el mundo” como punto de entrada para dispositivos importantes para la salud pública.

Con Donald Trump, quien ha prometido reducir las regulaciones, la FDA ha propuesto agilizar las pruebas previas a la comercialización de algunos dispositivos de alto riesgo. Esta medida podría recortar años de pruebas antes del lanzamiento de un producto y ahorrarles a las empresas millones de dólares. En un discurso a un grupo de la industria en mayo, Shuren reconoció que la política propuesta significaba “esencialmente aceptar un poco más de incertidumbre” en algunos casos.
En 2017 la FDA aprobó más del triple de dispositivos que en 2010, mientras que sus advertencias a los fabricantes de dispositivos sobre la seguridad de los productos se redujeron casi 80%.
La FDA le dijo a ICIJ que su objetivo de ser “el primero en el mundo” refleja la preocupación por los retrasos que impiden que las “nuevas tecnologías pioneras” lleguen a los pacientes estadounidenses más rápido que a los de otros países desarrollados, y que sigue comprometida a garantizar que esos dispositivos sean seguros y eficaces. Aunque la agencia está emitiendo menos cartas de advertencia, está llevando a cabo más inspecciones en las fábricas, afirmó la FDA.
Los críticos dicen que el tono de la FDA señala un cambio preocupante y se acerca hacia una estrategia promovida por la industria. “El tenor desregulador de los últimos años plantea preguntas reales sobre si la agencia es capaz de proteger adecuadamente al público estadounidense de dispositivos inseguros o ineficaces”, cuestionó el doctor Peter Lurie, ex comisionado asociado de la FDA.
El cabildeo de la industria de dispositivos médicos es una fuerza poderosa en Washington. Desde 2008, ha gastado más de 362 millones de dólares para influir en la legislación, según el Center for Responsive Politics. Los fabricantes también financian 35% del presupuesto del programa de dispositivos de la FDA a través de “cuotas de usuario” que se renegocian cada cinco años.
Estas cuotas le dan a la industria el poder de reformar a su regulador, según. Michael Carome, director del Public Citizen’s Health Research Group, un grupo de investigación sobre salud pública. El resultado de las negociaciones de cuotas “a menudo equivale al cumplimiento de una lista de deseos de la industria”, dijo Carome.
La formidable presencia de la industria en la agencia también se evidencia en las décadas de controversia sobre la seguridad de los implantes de mamas. Después de una virtual prohibición y una feroz lucha interna, la FDA permitió que la versión de silicona volviera al mercado en 2006, a pesar de la escasez de datos que mostraran la seguridad a largo plazo del producto.
“Fueron tenaces”, comentó Susan Wood, directora de la Oficina de Salud de la Mujer de la FDA entre 2000 y 2005. “A diferencia de otras compañías, después de que les dijeron que no, regresaron. Una y otra y otra vez. Desgastan cualquier resistencia”.
UNA RUPTURA DE LA CONFIANZA
Después de los reguladores, una última línea de defensa se interpone entre un paciente y un mal dispositivo: su médico. Pero esa línea también ha sido quebrantada.
Los cardiólogos, ortopedistas y otros médicos que implantan dispositivos están influenciados por una amplia gama de fuentes, incluyendo conferencias médicas, seminarios de capacitación y representantes de ventas de los fabricantes. Es común que éstos se reúnan en la sala de operaciones con los cirujanos, ofreciendo consejos sobre los dispositivos que venden.
En 2016, investigadores de la Universidad de Georgetown encontraron que los eventos patrocinados por las empresas y la presencia de sus empleados en los quirófanos socavan la independencia de los médicos y su capacidad para elegir el mejor tratamiento. Un administrador del hospital citado en el estudio describió la relación entre los cirujanos y los representantes de ventas como un “balde incestuoso de gusanos”.
Los médicos y los fabricantes caminan, en algunos casos, juntos en el negocio. Las empresas pagan regalías por las tecnologías desarrolladas con médicos y les otorgan subvenciones para investigar y opciones de compra de acciones de la empresa fabricante, lo que crea un conflicto de intereses que ha llamado la atención de las autoridades gubernamentales con frecuencia.
En Estados Unidos, donde los fabricantes de medicamentos y dispositivos están obligados a revelar los pagos que efectúan a los médicos, las 10 compañías más grandes de dispositivos médicos pagaron casi 600 millones de dólares a los médicos o a sus hospitales en 2017, según un análisis de ICIJ a partir de los datos de los centros de servicios de Medicare y Medicaid. Esta cifra no incluye los pagos relacionados con dispositivos de los gigantes que venden otros productos, como Johnson & Johnson y Allergan.
Un cirujano ortopédico de Los Ángeles, Thomas Schmalzried, ganó casi 30 millones de dólares en regalías y otros pagos de Johnson & Johnson por su papel en el diseño de dos sistemas de reemplazo de cadera de metal sobre metal, uno de los cuales fue retirado en todo el mundo ante la preocupación de que desprendiera niveles peligrosos de iones metálicos.
Schmalzried no respondió a las solicitudes de comentarios hechas por ICIJ. Johnson & Johnson dijo que aunque Schmalzried recibía regalías, no recibía nada por los productos que usaba en su consultorio o que eran implantados en hospitales donde tenía privilegios. La compañía informó que sus políticas prohíben tales pagos.
Las compañías de dispositivos han canalizado dinero, en ocasiones a través de cuentas en el extranjero, a distribuidores externos que luego pagan a cirujanos o a falsas organizaciones sin fines de lucro que los médicos crean para recibir los pagos, de acuerdo con documentos de las fiscalías de Estados Unidos e Italia.
Después de una serie de escándalos que condujeron a que se aprobara la Physician Payments Sunshine Act de 2010, una ley estadounidense que obligó a revelar estos pagos, la asociación comercial de la industria de dispositivos revisó su código de ética. El grupo propuso que se otorgara sólo una compensación “modesta” y “razonable” a los médicos que participan en eventos patrocinados por la empresa y establecer restricciones sobre las regalías y los acuerdos de consultoría.
Durante la última década, la asociación comercial europea ha acrecentado su código ético de 15 a 61 páginas, e incluso ha incorporado advertencias dirigidas a las empresas sobre las “posibles percepciones adversas del público” derivadas de los lugares en los que se realizan los eventos patrocinados. “Los cruceros, los clubes de golf o los balnearios y los lugares famosos por sus instalaciones de entretenimiento no son lugares apropiados”, establece el código.
Sin embargo, los funcionarios encargados de hacer cumplir la ley han seguido acusando a las compañías de dispositivos. Los médicos, los miembros de alto rango de la empresa y las autoridades gubernamentales han alegado en los tribunales sobre un hecho: los representantes de ventas influyen en las decisiones clínicas de los cirujanos y los alientan a utilizar los productos de maneras no aprobadas.
Tanto las empresas de la lista Fortune 500, por ejemplo Medtronic, como firmas más pequeñas de la industria han sido objeto de tales acusaciones.
En 2014, Biotronik, fabricante alemán de dispositivos médicos, pagó 4.9 millones de dólares para dejar fuera de juicio las acusaciones presentadas por el Departamento de Justicia de Estados Unidos, según las cuales pagaba sobornos a los médicos y promocionaba ilegalmente dispositivos cardiacos que no estaban autorizados por las autoridades.
Los representantes de ventas de la compañía recompensaban a los médicos que promovían terapias no autorizadas e implantaban un gran número de dispositivos Biotronik con entradas para eventos deportivos, salidas a campos de golf y lujosas comidas, según Brian Sant, un empleado de Biotronik que se convirtió en denunciante y cuya demanda provocó la investigación del gobierno. “Es casi como una anualidad”, escribió un gerente de ventas en un correo electrónico citado en la queja de Sant, refiriéndose a los pagos que los médicos podían recibir por inscribir a los pacientes en estudios patrocinados por la compañía.
En una respuesta escrita a ICIJ, Biotronik aseguró que “sus prácticas eran legales y éticas”. También dijo que el gobierno no investiga ninguna de las acusaciones sobre las prácticas de capacitación o los programas educativos de Biotronik.
Diversos gobiernos de países de todo el mundo también han acusado a los fabricantes de pagar a los administradores de hospitales y a los médicos con relojes Armani y lujosos paquetes vacacionales para aumentar las ventas y asegurar los contratos. En México, los empleados de una empresa encargados de sobornar a los médicos tenían una palabra clave para referirse a los pagos ilícitos: chocolates.
Un representante de ventas de implantes de Johnson & Johnson está siendo juzgado en Italia por sobornar a un prominente cirujano de Milán con más de 20 mil dólares, costosos viajes, cenas y más dádivas para él y su hijo, a cambio de que el cirujano implantara articulaciones artificiales de J&J y promocionara su marca en programas de televisión. La empresa respondió que no podía referirse a un caso abierto, pero agregó que “ha cooperado completamente con la investigación”.
En respuesta a ICIJ, Matt Wetzel, abogado general asociado de AdvaMed, dijo que la industria está “dedicada a hacer negocios de la manera correcta, y las compañías de tecnología médica han invertido incontables recursos, tanto de capital como humanos, en el desarrollo de programas de cumplimiento de vanguardia”.
DAÑO OCULTO
Con la reducción de las barreras para que los nuevos implantes lleguen al mercado, una mayor responsabilidad se desplaza hacia la vigilancia de los problemas y la información a los pacientes cuando éstos surgen.
Estados Unidos tiene, con mucho, la mayor cantidad de datos públicos sobre los implantes que mutilan y matan. La FDA lo mantiene en la base de datos “Manufacturer and User Facility Device Experience”, conocida como Maude.
Pero Maude depende de las propias compañías de dispositivos para recibir la gran mayoría de los datos. Por ley, los fabricantes deben recopilar las quejas de médicos, hospitales, pacientes, abogados y otros, y transmitirlas a la FDA.
En los hechos, las empresas de dispositivos suelen proporcionar información errónea o engañosa, o no la reportan en absoluto.
Entre 2008 y 2018, los inspectores de la FDA denunciaron a los fabricantes más de 4.400 veces por infringir su política de manejo de quejas. Cada infracción puede cubrir cientos o incluso miles de quejas mal manejadas, perdidas o enterradas.
La FDA reportó a Philips Healthcare, con sede en Cleveland, Ohio, por el mal manejo de miles de quejas sobre equipos de imagenología médica que revelaron problemas de alto riesgo que podrían causar lesiones o incluso muertes, incluyendo reportes de que los escáneres corporales habían mezclado imágenes de los pacientes. En las respuestas escritas a ICIJ, Philips señaló que ningún paciente resultó perjudicado en los incidentes y que su equipo de revisión de registros había evaluado todas las quejas.
Cuando las compañías reportan los incidentes adversos, a veces ocultan su gravedad. ICIJ descubrió que los fabricantes han clasificado un evento como “mal funcionamiento” o “lesión” aunque el paciente haya muerto.
Al utilizar un algoritmo de aprendizaje automático (machine learning) para buscar entre millones de informes, ICIJ encontró 2.100 casos en los que murieron personas, pero sus muertes se clasificaron como “averías” o “lesiones”. Del total, 220 informes mostraron que los dispositivos pueden haber causado o contribuido a las muertes. Los otros reportes no incluyeron suficiente información para determinar de manera concluyente si el dispositivo jugó un papel en la muerte de los pacientes.
Las directrices de la FDA exigen a los fabricantes que informen sobre las muertes que puedan estar relacionadas con sus dispositivos, incluso si la vinculación es incierta.
La FDA utiliza los informes de incidentes adversos para ayudar a identificar dispositivos peligrosos. “Si una muerte se clasifica como un mal funcionamiento del dispositivo, es posible que nunca se detecte”, señaló Madris Tomes, ex especialista en datos de la FDA y actual directora de una empresa dedicada a analizar incidentes adversos.
Jacob Shani, presidente de cardiología del Centro Médico Maimónides de Brooklyn, Nueva York, dijo que la información sobre incidentes adversos proporcionada por los fabricantes y los médicos es esencial para decidir qué producto implantar. “Si no hay transparencia y honestidad, entonces olvídalo”, concluyó Shani.
SISTEMA DE ALARMA DESCOMPUESTO
Los informes que finalmente llegan a los reguladores pueden desencadenar una alerta de seguridad o un retiro del mercado. Estas instancias pueden exigir que los hospitales retiren los dispositivos de su inventario o, dependiendo de la gravedad, incluso dar inicio a una ola de cirugías para retirar los dispositivos a los pacientes.
El que un producto sea restringido o retirado del mercado depende del lugar donde uno viva, encontró ICIJ. Durante 2017 las autoridades sanitarias de Nueva Zelanda, Irlanda, Escocia e Inglaterra restringieron fuertemente el uso de la malla vaginal utilizada para tratar la incontinencia o para mantener en su lugar los órganos reproductivos mientras los reguladores evaluaban la seguridad de los dispositivos. Pero esa malla se sigue vendiendo abiertamente en el mercado en otros países, entre ellos Canadá y Sudáfrica, donde Renate Scheepers recibió la suya.
Las alertas de seguridad supervisadas por el gobierno, a menudo llamadas “retiradas” (“recalls”) en Estados Unidos o “avisos de seguridad en el campo” en otros lugares, pueden variar desde simples cambios de etiqueta hasta la retirada inmediata del mercado. En algunos casos, los fabricantes retiran los productos discretamente sin admitir la culpa.
Los expertos dijeron a ICIJ que los gobiernos deben emitir avisos de retirada y alertas de seguridad para que los pacientes y los médicos estén al tanto de los problemas. Un análisis del ICIJ reveló que algunos gobiernos publican avisos con frecuencia y otros casi nunca lo hacen. Los reguladores sanitarios de México sólo compartieron información sobre dos retiros de productos. En Estados Unidos, la FDA ha publicado más de 26 mil en la última década.
Una cadera de metal sobre metal fabricada por Biomet, con sede en Indiana, se ha vinculado a la metalosis que provoca putrefacción del tejido. La compañía suspendió las ventas de este dispositivo hace varios años. Posteriormente, Biomet envió alertas de seguridad a cirujanos y otros proveedores de servicios médicos en Australia, Reino Unido y muchos otros países de Europa Occidental en 2015 y 2016, pero no a los de Canadá y Estados Unidos. Si la FDA hubiera presionado por el retiro de las caderas de Biomet en EE.UU., la compañía podría haberse visto forzada a enviar cartas de ese tipo a doctores de ese país.
“Adherimos a estrictas normas regulatorias y trabajamos en estrecha colaboración con la FDA y todas las agencias reguladoras pertinentes en cada una de nuestras regiones como parte de nuestro compromiso de operar un sistema de gestión de calidad de primera clase en toda nuestra red de fabricación global”, afirmó en un comunicado la compañía, ahora llamada Zimmer Biomet.
En una declaración enviada a ICIJ, la FDA apuntó a una comunicación generalizada de seguridad que posteó online en 2011 acerca de caderas de metal sobre metal como la razón para no exigir el retiro de los dispositivos de Biomet.
Los funcionarios a menudo no pueden encontrar a los pacientes con dispositivos defectuosos, ni siquiera a los médicos que los implantaron. Harold Paz, director médico y vicepresidente ejecutivo de Aetna, una de las aseguradoras de salud más grandes de Estados Unidos, destacó el historial comparativamente bueno de la industria automotriz para comunicarse con los propietarios de automóviles retirados del mercado.
“No tenemos manera de identificar cuál de nuestros miembros ha recibido el implante afectado”, dijo Paz.
Los pacientes que vivían con dispositivos que ya habían sido retirados del mercado dijeron que no se les había informado de los problemas aun después de la retirada. En entrevistas con los socios de ICIJ, cientos de receptores de implantes dijeron que los médicos nunca les advirtieron sobre los riesgos y no les informaron sobre las retiradas del mercado ni sobre las alertas de seguridad.
Connie Hill, una residente de 72 años de Sun City, Arizona, es una de varias pacientes que dicen que desearían haber sabido antes de las alertas de seguridad en el extranjero del implante de cadera Biomet. “Nunca oí nada al respecto”, le dijo Hill a ICIJ.
REPARAR UN “SISTEMA ROTO”
A principios de la década de 1990, un cirujano ortopédico australiano llamado Stephen Graves estaba cada vez más preocupado por los dispositivos médicos que implantaba. Los reemplazos de cadera y rodilla conllevaban grandes beneficios para los pacientes y también grandes riesgos, pero no tenía ni idea qué dispositivo era más seguro que otro. “No sabíamos cuántos o qué tipos de dispositivos estaban entrando”, comentó Graves. “Y no sabíamos el desempeño comparativo de los dispositivos.”
Graves comenzó a trabajar en 1996 con un grupo de colegas para establecer una base de datos central que permitiera rastrear a los australianos con implantes de cadera y rodilla y monitorear su salud. En pocos años, el registro nacional de dispositivos de Graves tenía ubicados a la gran mayoría de los reemplazos de cadera y rodilla en Australia y revelaba docenas de dispositivos deficientes.
En 2009, los reguladores australianos usaron los datos de Graves para plantear las primeras inquietudes sobre la seguridad del ASR XL de Johnson & Johnson, la marca de cadera de metal sobre metal implantada a Vijay Vojhala, de Mumbai. Hasta la fecha, el registro ha identificado más de 150 productos de reemplazo de articulaciones con un desempeño deficiente, dijo Graves.
Un mejor sistema para rastrear los dispositivos después de que salen al mercado goza de un amplio apoyo entre la industria, los médicos y los defensores de los pacientes. Henrik Malchau, cirujano ortopédico sueco que ha ayudado a establecer varios registros nacionales, dijo que éstos permiten a las autoridades alertar a los médicos y pacientes. En referencia al caso australiano, dijo: “Lo mejor de todo era que podían consultar el registro y encontrar a todos los pacientes”.
Después de los recientes retiros de dispositivos de gran repercusión mediática, India está evaluando una propuesta para crear su propio registro de reemplazo de articulaciones, mientras que otras iniciativas avanzan en Finlandia, Noruega y el Reino Unido, dijo Malchau.
Estados Unidos sigue estando rezagado. En un esfuerzo por mejorar la vigilancia post-implante de los dispositivos, la FDA está tratando de unificar en un solo sistema centralizado a varios pequeños registros privados con datos de reclamos de seguros.
Este programa depende en gran medida del éxito de una iniciativa de la FDA que exige que a cada dispositivo se le asigne un número de identificación único para que sea más fácil de rastrear. Aunque un programa de “identificador único de dispositivos” (Unique Device Identifier, UDI) podría marcar el comienzo de una era de vigilancia poscomercialización más avanzada, su plena aplicación se encuentra a años de distancia. Un obstáculo potencial: el gobierno federal aún no ha emitido reglas finales que obliguen a incluir identificadores de dispositivos en los registros electrónicos de salud y en los datos de los reclamos de seguros, que son formas cruciales en las que la iniciativa puede ser utilizada para ayudar a rastrear a los pacientes y vigilar el desempeño de sus dispositivos.
Y la perspectiva de un sistema de UDI coordinado a nivel mundial —ahora en discusión entre los reguladores de dispositivos médicos— es todavía más lejana.
Hanifa Koya, ginecóloga de Wellington, Nueva Zelanda, que ha realizado muchas cirugías horripilantes para eliminar mallas defectuosas, dijo que es fundamental llevar un registro de los dispositivos vendidos e implantados. “Si los cirujanos están buscando constantemente implementar dispositivos innovadores pero no piden un registro, entonces éste es un sistema roto”, dijo.
Koya se mostró convencida de que los paneles de ética de los hospitales deberían tener el poder de prohibir a los médicos el uso de implantes que no hayan sido probados o sobre los que haya dudas en torno a su seguridad.
Pero reconoció que no será fácil que estos problemas se subsanen. E incluso que éstos son apenas una pequeña pieza de problemas sistémicos más grandes.
“Cuando se trata de proteger a la gente que sufrirá”, dijo Koya, “el sistema está absolutamente en ruinas”.
* Contribuyeron a esta historia: Ben Hallman, Jet Schouten, Dean Starkman, Simon Bowers, Emilia Díaz-Struck, Gerard Ryle, Sasha Chavkin, Spencer Woodman, Cat Ferguson, Petra Blum, Scilla Alecci, Sydney P. Freedberg, Fergus Shiel, Richard H. P. Sia, Tom Stites, Martha M. Hamilton, Joe Hillhouse, Rigoberto Carvajal, Cécile Schilis-Gallego, Hilary Fung, Marina Walker Guevara, Miguel Fiandor, Pierre Romera, Hamish Boland-Rudder, Will Fitzgibbon, Delphine Reuter, Amy Wilson-Chapman, Margot Williams, Pauliina Siniauer, Razzan Nakhlawi, Jesse McLean, Matthew Perrone, Holbrook Mohr, Mitch Weiss, Esther Oxford, Charles Babcock, Andrew Lehren y Emily Siegel.
Traducción: Alan Porter



